Abstract
A fully resolved consensus between fully resolved phylogenetic trees
Author(s): Jos├?┬® Augusto Amgarten Quitzau and Jo├?┬Żo MeidanisNowadays, there are many phylogeny reconstruction methods, each with advantages and disadvantages. We explored the advantages of each method, putting together the common parts of trees constructed by several methods, by means of a consensus computation. A number of phylogenetic consensus methods are already known. Unfortunately, there is also a taboo concerning consensus methods, because most biologists see them mainly as comparators and not as phylogenetic tree constructors. We challenged this taboo by defining a consensus method that builds a fully resolved phylogenetic tree based on the most common parts of fully resolved trees in a given collection. We also generated results showing that this consensus is in a way a kind of “median” of the input trees; as such it can be closer to the correct tree in many situations.
Impact Factor an Index

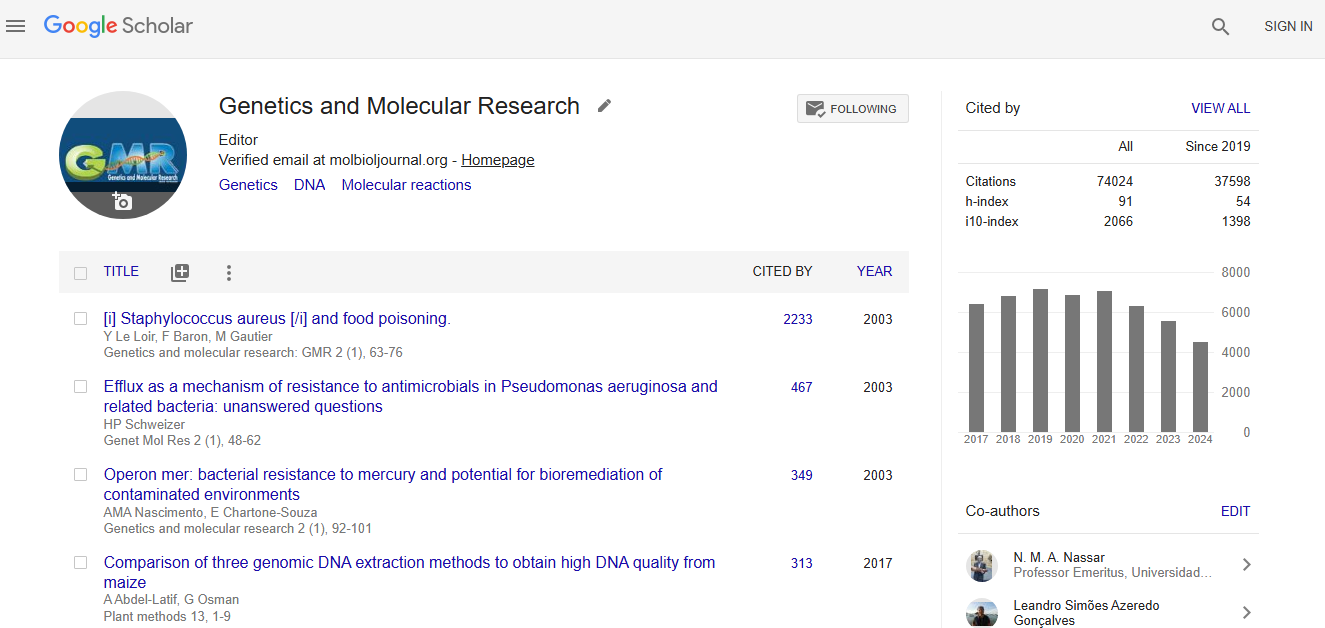
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report