Abstract
A novel stop codon mutation in exon 1 (558C>A) of the UGT1A1 gene in a Thai neonate with Crigler-Najjar syndrome type I
Author(s): N. Wanlapakorn, P. Nilyanimit, T. Vorawandthanachai, T. Deesudjit, N. Dumrongpisutikul and Y. PoovorawanHuman uridine 5'-diphosphate-glucuronosyltransferases play a critical role in detoxification by conjugating bilirubin with glucoronic acid. Impaired or reduced enzymatic activity causes a spectrum of clinical disorders such as Crigler-Najjar syndrome type I (CN1), Crigler-Najjar syndrome type II, and Gilbert��?�?�?s syndrome. CN1 is a severe form of unconjugated hyperbilirubinemia caused by homozygous or compound heterozygous mutations in the gene for uridine 5'-diphosphate glucuronosyltransferase 1 family, polypeptide A1 (UGT1A1), resulting in complete loss of enzyme function. Here, we report a novel homozygous mutation of UGT1A1 in a female Thai infant who was diagnosed with CN1, and her parents were found to be heterozygous carriers. The patient was homozygous for the c.558C>A mutation, which resulted in a premature stop codon in exon 1. Her asymptomatic parents were carriers of the nonsense c.558C>A mutation. Our result suggests an important role for homozygous c.558C>A mutations in the UGT1A1 gene in the development of severe unconjugated hyperbilirubinemia.
Impact Factor an Index

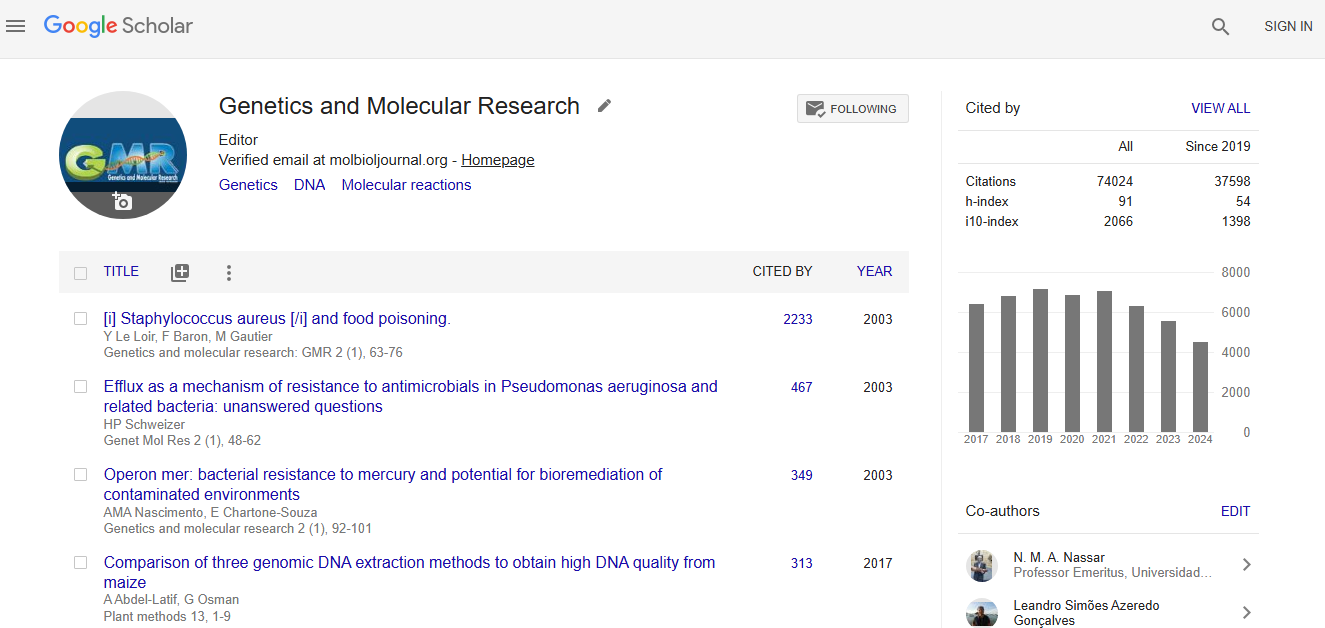
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report