Abstract
High-accuracy splice site prediction based on sequence component and position features
Author(s): J.L. Li L.F. Wang H.Y. Wang L.Y. Bai Z.M. YuanIdentification of splice sites plays a key role in the annotation of genes. Consequently, improvement of computational prediction of splice sites would be very useful. We examined the effect of the window size and the number and position of the consensus bases with a chi-square test, and then extracted the sequence multi-scale component features and the position and adjacent position relationship features of consensus sites. Then, we constructed a novel classification model using a support vector machine with the previously selected features and applied it to the Homo sapiens splice site dataset. This method greatly improved cross-validation accuracies for training sets with true and spurious splice sites of both equal and different proportions. This method was also applied to the NN269 dataset for further evaluation and independent testing. The results were superior to those obtained with previous methods, and demonstrate the stability and superiority of this method for prediction of splice sites. Identification of splice sites plays a key role in the annotation of genes. Consequently, improvement of computational prediction of splice sites would be very useful. We examined the effect of the window size and the number and position of the consensus bases with a chi-square test, and then extracted the sequence multi-scale component features and the position and adjacent position relationship features of consensus sites. Then, we constructed a novel classification model using a support vector machine with the previously selected features and applied it to the Homo sapiens splice site dataset. This method greatly improved cross-validation accuracies for training sets with true and spurious splice sites of both equal and different proportions. This method was also applied to the NN269 dataset for further evaluation and independent testing. The results were superior to those obtained with previous methods, and demonstrate the stability and superiority of this method for prediction of splice sites.
Impact Factor an Index

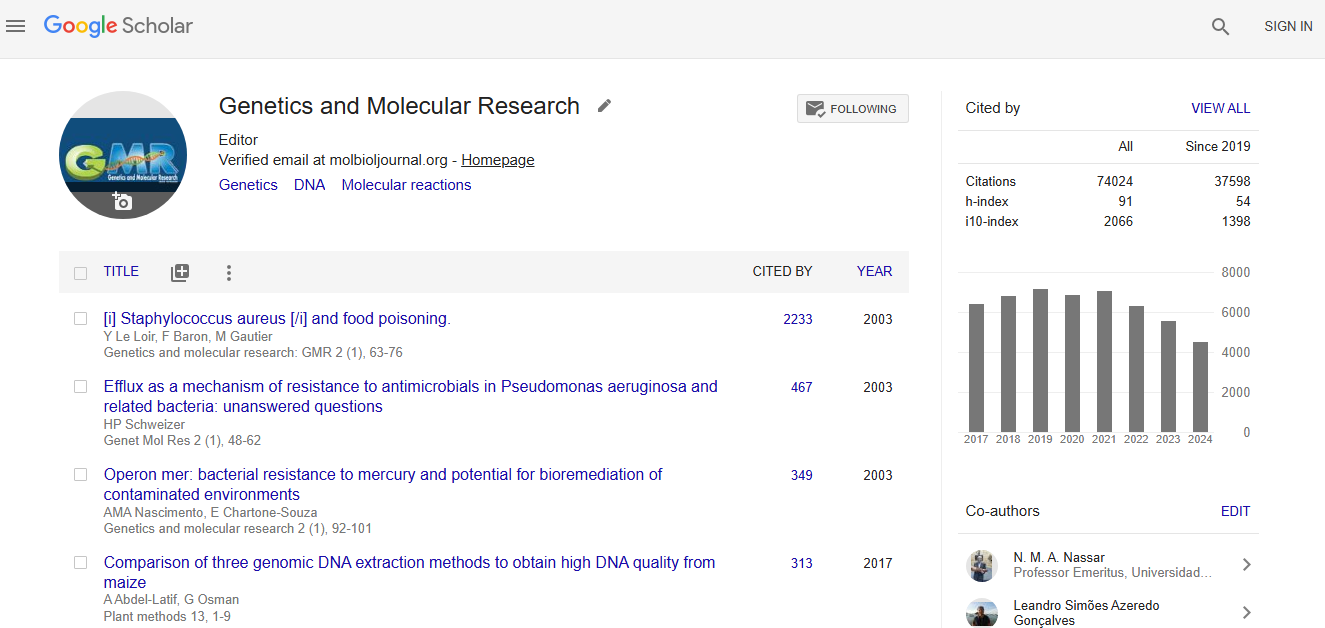
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report