Abstract
Identification and complete sequencing of novel human transcripts through the use of mouse orthologs and testis cDNA sequences
Author(s): Elisa N. Ferreira, Lilian C. Pires, Raphael B. Parmigiani,Fabiana Bettoni, Renato D. Puga, Daniel G. Pinheiro,LuÃ?Âs Eduardo C. Andrade, Luciana O. Cruz, Theri L. Degaki,Milton Faria Jr., Fernanda Festa, Daniel Giannella-Neto,Ricardo R. Giorgi, Gustavo H. Goldman, Fabiana Granja,Arthur Gruber, Christine Hackel, FlÃ?¡vio Henrique-Silva,Bettina Malnic, Carina V.B. Manzini, Suely K.N. Marie14,Nilce M. Martinez-Rossi15, Sueli M. Oba-Shinjo14,Maria Ines M.C. Pardini16, Paula Rahal18, ClÃ?¡udia A.Rainho,Silvia R. Rogatto, Camila M. Romano, Vanderlei Rodrigues,Magaly M. Sales, Marcela Savoldi8, Ismael D.C.G. da Silva,Neusa P. da Silva, Sandro J. de Souza, Eloiza H. Tajara,Wilson A. Silva Jr, Andrew J.G. Simpson, Mari C. Sogayar,Anamaria A. Camargo and Dirce M. CarraroThe correct identification of all human genes, and their derived transcripts, has not yet been achieved, and it remains one of the major aims of the worldwide genomics community. Computational programs suggest the existence of 30,000 to 40,000 human genes. However, definitive gene identification can only be achieved by experimental approaches. We used two distinct methodologies, one based on the alignment of mouse orthologous sequences to the human genome, and another based on the construction of a high-quality human testis cDNA library, in an attempt to identify new human transcripts within the human genome sequence. We generated 47 complete human transcript sequences, comprising 27 unannotated and 20 annotated sequences. Eight of these transcripts are variants of previously known genes. These transcripts were characterized according to size, number of exons, and chromosomal localization, and a search for protein domains was undertaken based on their putative open reading frames. In silico expression analysis suggests that some of these transcripts are expressed at low levels and in a restricted set of tissues.
Impact Factor an Index

Google scholar citation report
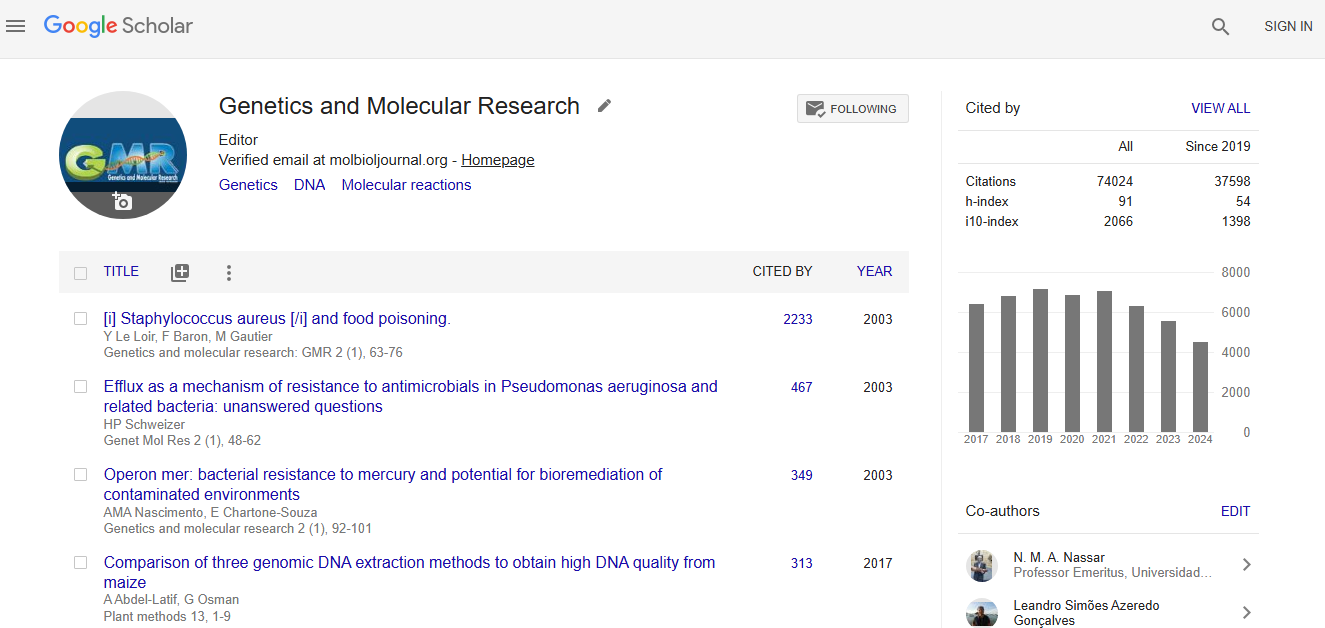
Citations : 56184
Genetics and Molecular Research received 56184 citations as per google scholar report