Abstract
Screening for key genes associated with invasive ductal carcinoma of the breast via microarray data analysis
Author(s): J.L. Huan, X. Gao, L. Xing, X.J. Qin, H.X. Qian, Q. Zhou and L. ZhuThe aim of this study was to identify key genes related to invasive ductal carcinoma (IDC) of the breast by analyzing gene expression data with bioinformatic tools. Microarray data set GSE31138 was downloaded from Gene Expression Omnibus, including 3 breast cancer tissue samples and 3 normal controls. Differentially expressed genes (DEGs) between breast cancer and normal control were screened out (FDR < 0.05 and |logFC| > 2). Coexpression between genes was examined with String, and a network was then constructed. Relevant pathways and diseases were retrieved with KOBAS. A total of 56 DEGs were obtained in the IDC of the breast compared with normal controls. A gene coexpression network including 27 pairs of genes was constructed and all the genes in the network were upregulated. Further study indicated that most of the genes in the coexpression network were enriched in ECM-receptor interaction (COL4A2, FN1, and HMMR) and nucleotide excision repair(CETN2 and PCNA) pathways, and that the most significantly related disease was autoimmune lymphoproliferative syndromes. A number of DEGs were acquired through comparative analysis of gene expression data. These findings are beneficial in promoting the understanding of the molecular mechanisms in breast cancer. More importantly, some key genes were revealed via gene coexpression network analysis, which could be potential biomarkers for IDC of the breast.
Impact Factor an Index

Google scholar citation report
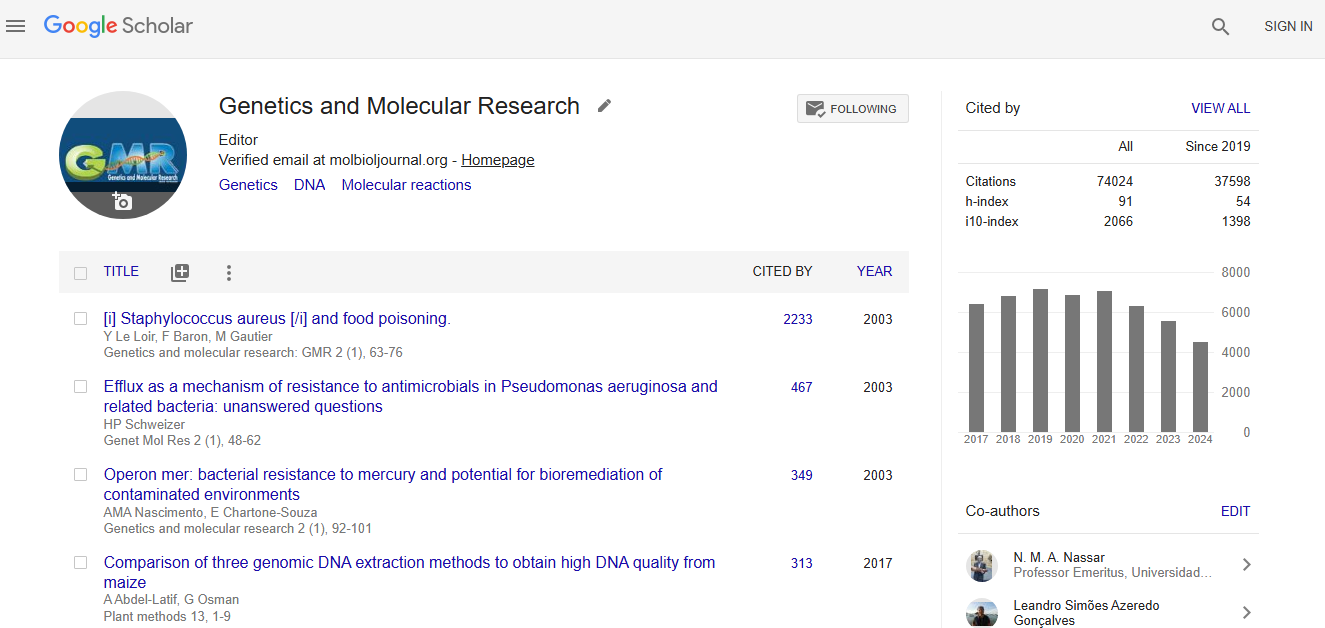
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report