Abstract
Using Markov chains of nucleotide sequences as a possible precursor to predict functional roles of human genome: a case study on inactive chromatin regions
Author(s): K.-E. Lee, E.-J. Lee, and H.-S. ParkRecent advances in computational epigenetics have provided new opportunities to evaluate n-gram probabilistic language models. In this paper, we describe a systematic genome-wide approach for predicting functional roles in inactive chromatin regions by using a sequence-based Markovian chromatin map of the human genome. We demonstrate that Markov chains of sequences can be used as a precursor to predict functional roles in heterochromatin regions and provide an example comparing two publicly available chromatin annotations of large-scale epigenomics projects: ENCODE project consortium and Roadmap Epigenomics consortium.
Impact Factor an Index

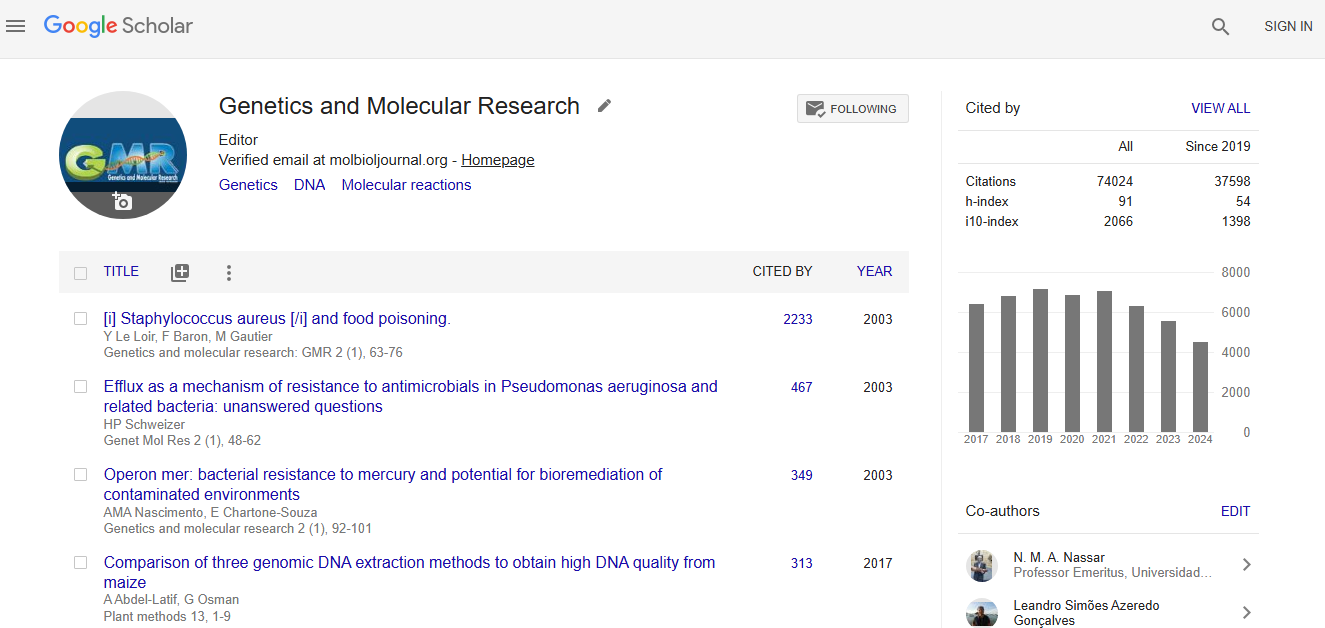
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report